AI in Pharma: Real-World Results From 1000+ Drug Discovery Projects
 AI is changing the pharmaceutical world. The market will grow from $3.24 billion in 2024 to $65.83 billion by 2033, with a CAGR of 39.74%. Drug development faces big challenges. A company needs about 10 years and $1.4 billion to bring just one drug to market. The timeline has dropped by around 18 months in the last two decades. The average cost keeps rising instead of falling.
AI is changing the pharmaceutical world. The market will grow from $3.24 billion in 2024 to $65.83 billion by 2033, with a CAGR of 39.74%. Drug development faces big challenges. A company needs about 10 years and $1.4 billion to bring just one drug to market. The timeline has dropped by around 18 months in the last two decades. The average cost keeps rising instead of falling.
AI tools speed up many parts of the drug discovery process. These tools can cut the discovery phase from 3-6 years down to just 1-2 years. AI helps predict how well drugs will work and how safe they are. It also makes clinical trials better. The results speak for themselves. AI-focused startups found 158 drug candidates between 2010 and 2022, and 15 of these moved to clinical trials. AI-powered drug discovery shows great promise. Phase 1 trials now succeed 80-90% of the time, which beats the usual industry rates of 40-65% by a lot. The McKinsey Global Institute believes AI in pharma and biotech could create $60-110 billion in economic value each year.
AI-Powered Target Identification Across 1000+ Projects
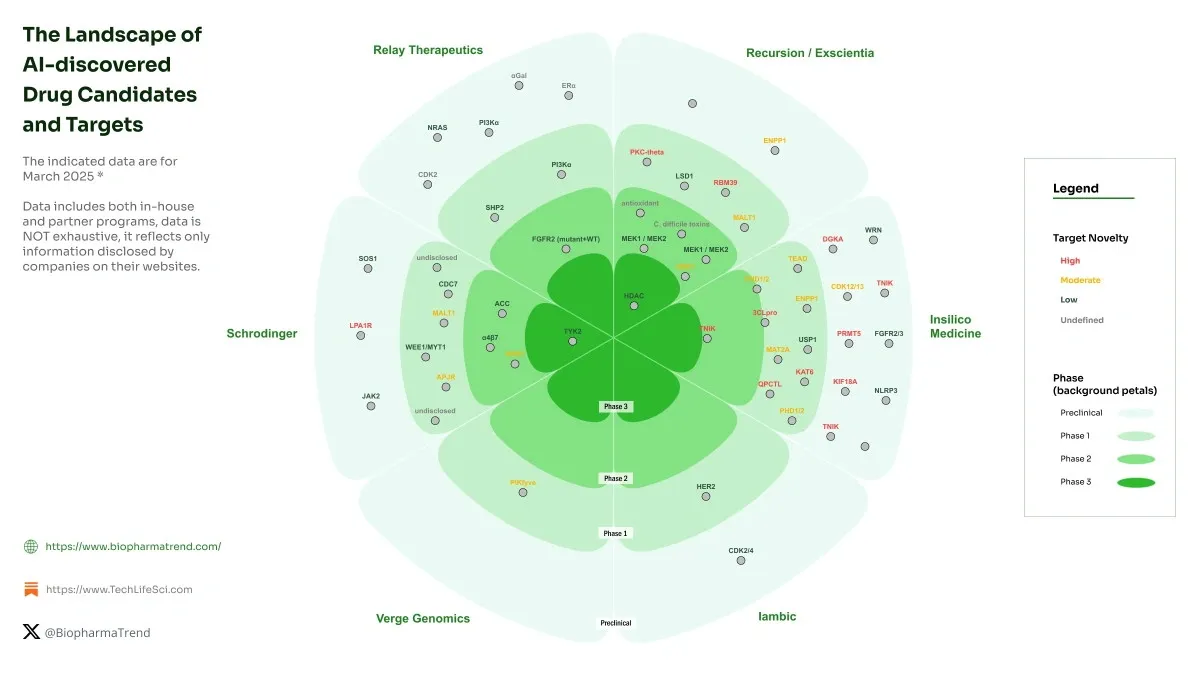
Image Source: BiopharmaTrend
Target identification sets the foundation for pharmaceutical research and leads to drug discovery processes. AI has become a game-changer that identifies viable therapeutic targets with unprecedented accuracy across more than 1,000 drug discovery projects. Evidence-based science thrives now because high-throughput technologies generate massive genomic datasets that AI systems analyze effectively.
Genomic Data Mining with Deep Learning
Deep learning extracts patterns from complex genomic data, particularly in the noncoding regions of the human genome—an untapped source of potential drug targets. Over 90% of pharmacogenomic SNPs associated with drug efficacy and adverse events exist within enhancers, promoters, and intron regions. This pharmacoepigenome’s regulatory elements and their variations account for more than 80% of genetic contributions to disease risk.
Scientists have developed several machine learning applications that predict noncoding SNPs’ effects. DeepSEA stands out as one of the first deep-learning frameworks that predicts chromatin effects of sequence alterations with single nucleotide sensitivity. DeepBind uses convolutional neural networks to calculate how well nucleotide sequences bind to transcription factors and helps characterize mutations’ effects on binding properties.
Target prioritization benefits from genome-wide association studies (GWAS) and phenome-wide association studies (PheWAS) data. The EMBL-EBI GWAS catalog contained associations from 5,370 studies and identified more than 290,000 associations as of October 2021. Tools like StarGazer combine this data with open-access databases to identify potential drug targets quickly.
Protein-Protein Interaction Mapping using Graph Neural Networks
Proteins work together with other proteins to complete biological activities. Scientists need to understand these protein-protein interactions (PPIs) to unravel cellular behavior and develop effective drugs. Graph neural networks have transformed PPI analysis methods.
Scientists represent proteins as graphs where each node works as an amino acid residue. Nodes connect when they have atoms within a threshold distance (typically 6 angstroms). Pre-trained protein language models process the protein’s sequence to extract features for each node.
Graph convolutional networks (GCNs) and graph attention networks (GATs) learn from these protein representations. Research on two PPI datasets—Human and S. cerevisiae—proved that graph-based approaches work better than previous leading methods. The Human dataset had 36,545 interacting protein pairs, while the S. cerevisiae dataset contained 17,257 pairs after preprocessing.
DeepGNHV has combined a pretrained protein language model with AlphaFold2’s structural features. It uses graph attention networks to predict human-virus PPIs with superior performance. This approach showed exceptional generalization capabilities with viral proteins absent from the training process.
Case Study: AstraZeneca’s Genome-Wide Association AI Pipeline
AstraZeneca’s Center for Genomics Research shows how AI works in target identification. The center aims to analyze up to two million genomes by 2026. Cloud environments and advanced AI tools help the company process vast genomics data faster and more effectively than before.
MILTON (MachIne Learning with phenoType associatiONs), an advanced machine learning research tool, predicts more than 1,000 diseases before diagnosis. Using data from nearly 500,000 UK Biobank participants, MILTON achieved:
-
High predictability for 1,091 diseases (area under curve above 0.7)
-
Exceptional performance for 121 diseases (AUC above 0.9)
MILTON helps researchers identify individuals who might have been incorrectly classified as controls in conventional case-control studies. This improves the statistical power for genetic discovery. AI-increased reclassification expands gene discovery’s scope and accuracy for hundreds of diseases.
AstraZeneca added the first two AI-generated drug targets to their portfolio through collaboration with BenevolentAI in 2021. Knowledge graphs integrate genomic, disease, drug, clinical, and safety information to overcome confirmation bias and turn data into insights.
Virtual Screening and Hit Discovery at Scale

Image Source: MDPI
Virtual screening has become the life-blood of modern drug discovery. Scientists can now test millions of compounds without spending time and money on physical testing. These computer techniques have sped up early drug development in more than 1,000 pharmaceutical projects. Let’s get into how advanced AI technologies are changing this key phase.
Atomwise’s Structure-Based Screening Engine
Atomwise’s breakthrough AtomNet platform is a big step forward in virtual high-throughput screening (HTS). A study with 318 targets through collaboration with more than 250 academic labs in 30 countries showed amazing results. AtomNet had a 74% success rate. This rate was way higher than the usual 50% seen in traditional HTS approaches. This breakthrough proved virtual screening could work just as well as old methods.
AtomNet’s real power lies in its ability to search through more than 15 quadrillion compounds that can be made. Traditional HTS can’t test 99% of compounds available in the market. But AtomNet quickly explores these big, untested chemical spaces.
The platform finds seven different bioactive compounds for each target. This solves a basic problem in finding small molecule drugs. Scientists can now find new molecular frameworks that might work better for selectivity, pharmacokinetics, or pharmacodynamics.
AtomNet does all this without needing target-specific training data. Scientists can use it for any target, even ones they thought were impossible to drug before. This marks a fundamental change from target-specific models to a global, pre-trained approach that works across the proteome.
Support Vector Machines in Ligand-Target Prediction
Support vector machines (SVMs) are now key to virtual screening. They’re great at classifying compounds and predicting activity. SVMs started as the go-to method in chemoinformatics. They’re really good at predicting compound class labels—exactly what virtual screening needs.
SVMs work by using kernel functions. These functions map problems into spaces where they’re easier to solve. The radial basis function (RBF) kernel does this mapping in a non-linear way. Scientists have used it with great results in many QSAR studies. For ligand prediction, SVM calculations mix protein and small molecule data through special target-ligand kernel functions.
These kernels look at different types of target information:
-
Sequence, secondary, and tertiary structures
-
Biophysical properties
-
Ontologies and structural taxonomy
SVMs work well even with orphan targets (targets without known ligands). The models can predict ligands accurately if they have compounds from similar targets to learn from. SVR (Support Vector Regression) has also become a main tool for non-linear QSAR and virtual screening.
AlphaFold Integration for Binding Site Prediction
Adding AlphaFold to virtual screening is another huge leap forward in finding new drugs. AlphaFold models show binding pocket structures with amazing accuracy. The errors are almost as small as differences between lab-tested structures of the same protein with different bound ligands.
AlphaFold 2 (AF2) models work much better than old homology models. AF2 models have a median RMSD of 1.3 Å, while traditional template-based models show 3.3 Å. This accuracy is getting close to lab-tested structures, making them more valuable for finding new drugs.
In spite of that, some problems still exist. Ligand binding poses from AF2 models weren’t much better than traditional models (16% versus 11% correct poses). Both performed worse than lab-tested protein structures (48%). This shows that even with better structural accuracy, predicting how ligands and proteins interact remains tricky.
The OpenVS platform combines AI-powered virtual screening with everything needed to find new drugs. This scalable, open-source platform can test billions of chemical compounds against target proteins in just seven days using powerful computers.
These technologies keep getting better. Together, they’re changing how we find new drug candidates, making the process faster and cheaper.
Lead Optimization Using Predictive Modeling
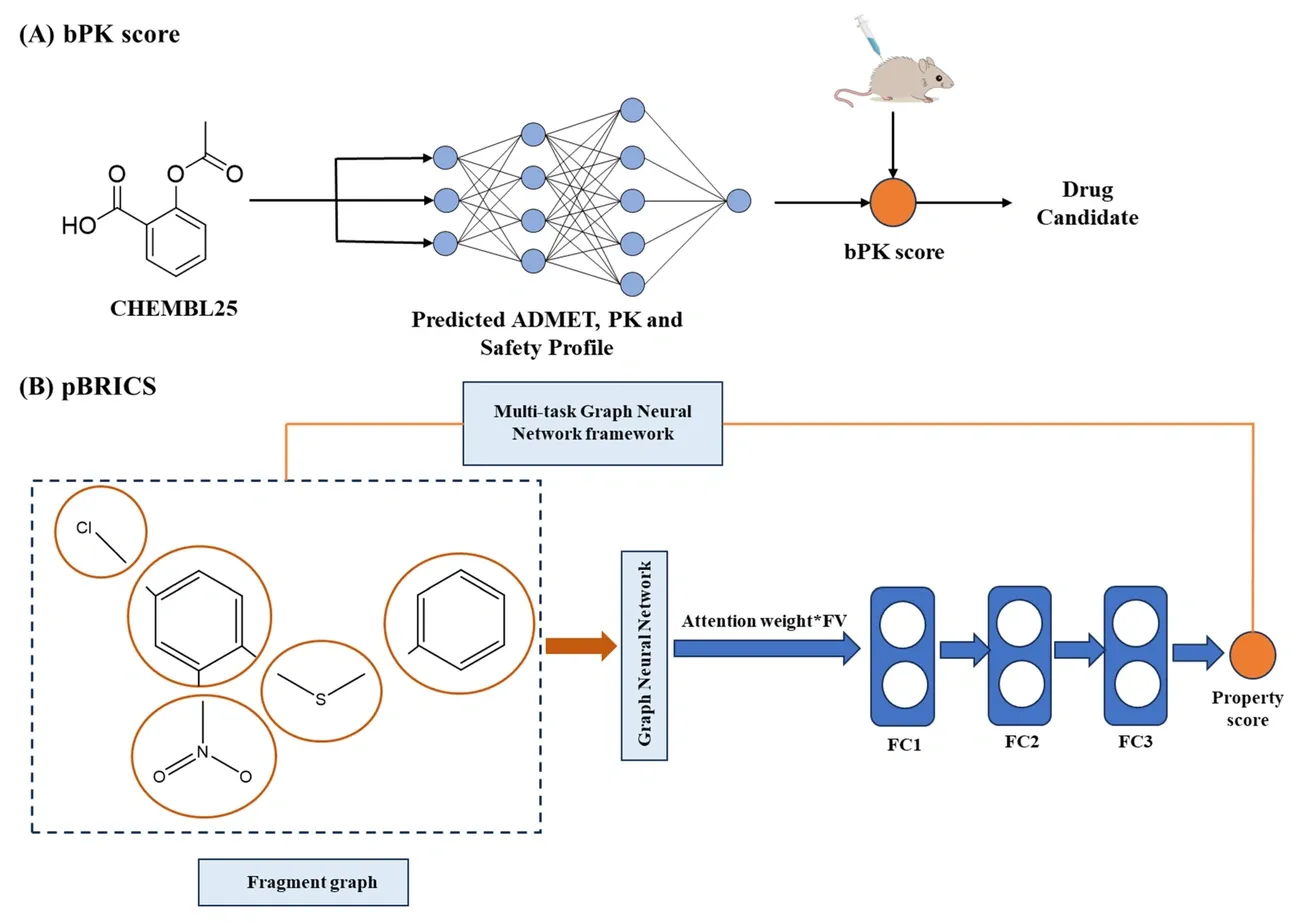
Image Source: MDPI
Drug developers face a key challenge after they find potential candidates: making these molecules better in terms of potency, selectivity, and pharmacokinetic properties. AI technologies are proving valuable in lead optimization by cutting down the time and resources needed to bring drugs to market.
QSAR Modeling for ADME Profiling
ADME (absorption, distribution, metabolism, and excretion) problems cause many clinical failures in drug development. QSAR modeling helps solve this by connecting molecular structures to their biological properties. These models work on a basic principle: similar chemical structures show similar biological activities.
Today’s QSAR approaches employ molecular descriptors like weight, electronegativity, and hydrophobicity to capture key structural features that affect biological activity. The National Center for Advancing Translational Sciences (NCATS) has built successful QSAR models for three key ADME endpoints: kinetic aqueous solubility, parallel artificial membrane permeability, and rat liver microsomal stability. Tests against marketed drugs showed these models reached balanced accuracies ranging from 71% to 85%.
NCATS has made these prediction tools available to everyone through the ADME@NCATS web portal. This gives drug discovery researchers valuable resources. Scientists can now predict ADME properties early on and reduce their project’s risk profile.
Generative Adversarial Networks for Molecule Design
GANs have transformed molecule design by creating new compounds that meet specific biological and safety requirements. A GAN works with two competing neural networks: one creates new molecular structures based on existing beneficial compounds, while the other checks these structures against real molecules.
The generator gets better at creating realistic molecular structures as training progresses. Meanwhile, the discriminator becomes better at spotting artificial samples. This competition leads to new molecules with optimized properties.
Recent breakthroughs include:
-
MedGAN, which uses an optimized Wasserstein GAN with Graph Convolutional Networks to handle graph-structured data showing bonds between atoms
-
LatentGAN, which pairs an autoencoder and GAN for de novo drug design, solving the differentiation problem in discrete SMILES data
-
QuMolGAN, a hybrid quantum-classical GAN that gets better physicochemical properties with fewer learnable parameters than classical versions
Tests show MedGAN created compounds that matched Lipinski’s Rule of 5 in 96.1-99.2% of cases, and achieved synthetic accessibility scores below 6 for 96-100% of compounds. Better yet, 22-31% of the produced molecules showed no toxicity across 12 predefined targets in a pre-trained Tox21 model.
DeepChem Framework for Activity Prediction
DeepChem stands out as a python-based, open-source platform built specifically for drug discovery. Unlike older frameworks such as scikit-learn, DeepChem speeds up computation with NVIDIA GPUs and uses Google TensorFlow along with scikit-learn for neural networks.
DeepChem’s strength lies in its many publicly available chemical assay datasets. Performance assessment uses the squared Pearson Correlation Coefficient for regression datasets and the area under curve (AUC) for receiver operator characteristic (ROC) curves in classification datasets.
The platform has multiple ways to featurize molecules. SMILES strings give unique molecular representations, but most machine learning methods need more feature information. DeepChem solves this through various featurization techniques.
Scientists used DeepChem to predict molecular toxicity using the Toxicology in the 21st Century (Tox21) dataset. This dataset contains measurements of stress response and nuclear receptor pathway activation from 8,014 different molecules across 12 response pathways linked to toxicity. DeepChem’s deep learning algorithms learn molecular representations directly from chemical structures to predict new compound analogs’ biological activities with high accuracy.
Traditional drug development costs about $2.80 billion per approved drug with only a 2% success rate. This makes AI-powered predictive modeling crucial for pharmaceutical breakthroughs. These technologies make the lead optimization process more efficient by systematically exploring the big chemical space and accurately predicting drug properties.
AI in Preclinical and Clinical Trial Design
Clinical trials are the most expensive and time-consuming phase of drug development. The failure rates exceed 90% which makes this a big deal. AI in pharma is changing how researchers design and run these vital studies.
Patient Stratification with Ground Data
Patient populations’ diversity creates a huge challenge in clinical trials. AI provides solutions through precise stratification. Psychiatric trials don’t deal very well with biomarker identification. AI-enabled stratification tools match treatments to biological profiles before enrollment starts. The TAMARIND study shows this approach by using a genetic test (CRHR1CDx). This test identifies depression patients with specific HPA axis dysregulation and enrolls only those who will likely benefit from the treatment.
AI has improved stratification in gastrointestinal cancers by combining multiple data types. These include imaging, pathology, clinical variables, and molecular profiles. The KEYNOTE-062 study utilized an AI model that included tumor mutational burden. This model showed stronger correlation with clinical outcomes in gastric cancer patients receiving pembrolizumab and performed better than traditional stratification methods.
Predictive Toxicology using Ensemble Models
Toxicity issues are the main reasons clinical trials fail. AI-powered models now identify toxicity risks early by utilizing large-scale datasets. These include omics profiles, chemical properties, and electronic health records. This approach reduces animal testing and aligns with the 3Rs principle (Replacement, Reduction, and Refinement).
Scientists developed an optimized ensemble model that combines random forest and k-star techniques. This model achieved 93% accuracy in toxicity prediction. The accuracy was 8% higher than the best single machine learning model and 21% better than leading deep learning approaches. Principal component analysis for feature selection helped the model show remarkable improvement across all evaluation parameters.
Trial Simulation with Bayesian Optimization
Bayesian optimization helps researchers select optimal clinical trial designs when they face numerous parameter options. This approach streamlines processes to identify parameters that maximize statistical power within fixed sample size constraints, especially for complex designs without analytical solutions.
Pre-trial simulations of 10,000 trials across 540 adaptive designs helped researchers find optimal adaptation settings. These simulations totaled 5.4 million simulated trials. The optimal Bayesian adaptive design with informative prior reduced sample size by 71% compared to traditional frequentist approaches. This meant using 198 participants instead of 682 while maintaining scientific validity.
These AI-powered approaches solve key challenges in modern clinical trials. They improve patient selection precision, enhance safety assessments, and optimize study designs efficiently without compromising scientific integrity.
Formulation and Drug Delivery Innovations
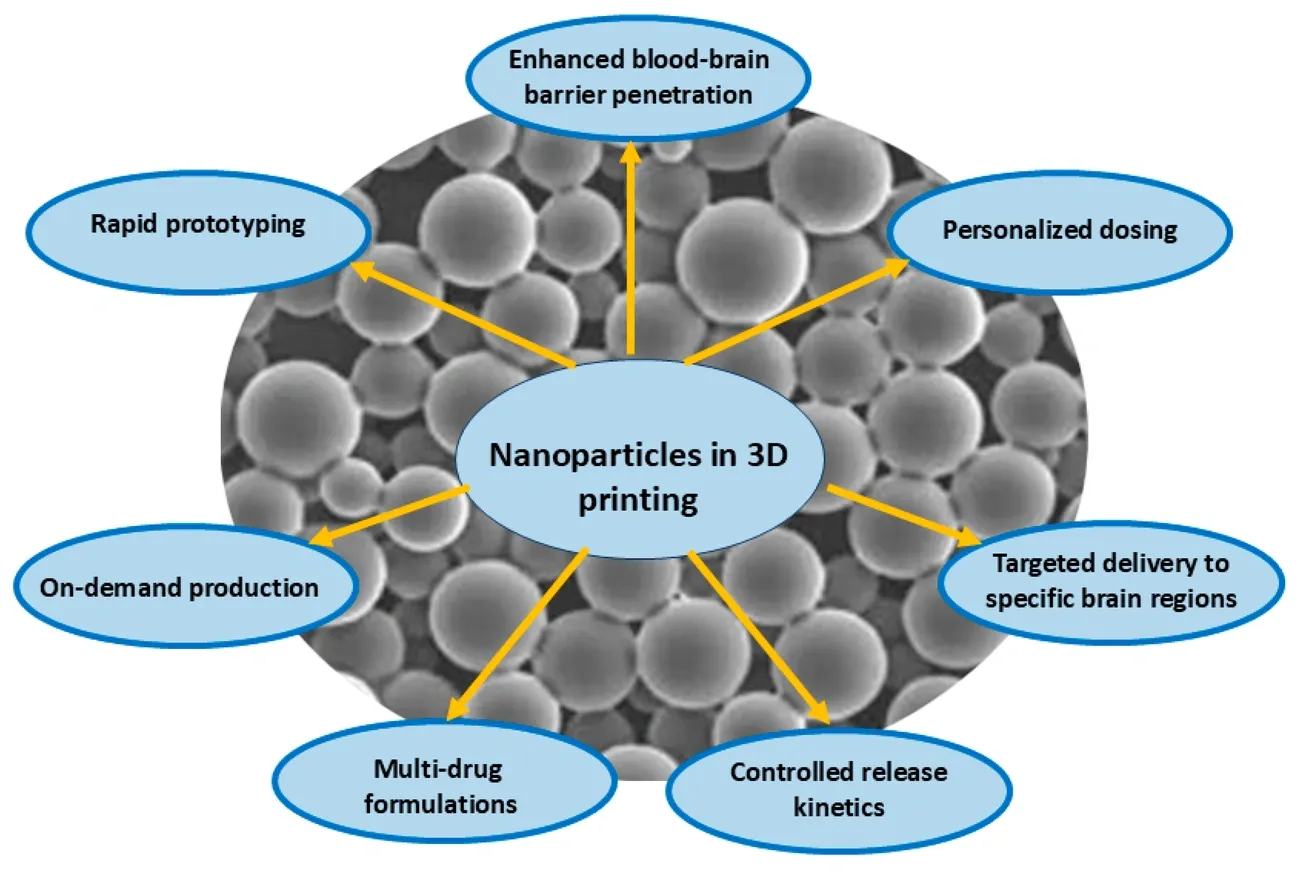
Image Source: MDPI
Pharmaceutical formulation development has traditionally required time-consuming experimental processes that combine active ingredients with excipients to create effective drug products. AI technologies now streamline this final yet significant phase of the drug development pipeline.
Excipient Compatibility Prediction with DE-INTERACT
Drug-excipient compatibility forms the foundation of pharmaceutical formulation stability. Machine learning powers the DE-INTERACT model to analyze molecular fingerprints of compounds and predict potential incompatibilities. This innovative tool uses 881-bit binary fingerprints of each drug and excipient, which creates 1,762 inputs per instance. The model achieved impressive training and validation accuracies of 99.3% and 91.6% respectively when tested on a dataset of over 3,500 drug-excipient pairs from peer-reviewed literature. DE-INTERACT successfully identified incompatibilities between paracetamol with vanillin, paracetamol with methylparaben, and brinzolamide with polyethyleneglycol. Conventional analytical techniques later confirmed these predictions.
AI-Driven Nanocarrier Design for Targeted Delivery
AI integration with nanoarchitectonics has created a transformation in targeted delivery system development. Machine learning helps optimize drug loading capacity, predict release kinetics, and improve targeting capabilities. Scientists use rational design, formulation, detailed characterization, and biological verification to generate multi-dimensional datasets that continuously refine predictive models.
AI-powered nanosystems show remarkable versatility in platforms of all types. Lipid-based carriers excel at drug encapsulation while AI models optimize membrane dynamics. Polymeric nanocarriers provide tunable degradation profiles that machine learning optimizes for precise spatiotemporal release. These smart multifunctional nanocarriers respond to specific triggers like pH changes, temperature, light, or enzymatic activity. This response enables controlled therapeutic cargo delivery with minimal premature elimination.
3D-Printed Dosage Forms Tailored by AI
Three-dimensional printing offers a groundbreaking approach to develop personalized oral dosage forms with complex geometric structures. AI optimization of printing parameters ensures quality control and efficiency. Machine learning algorithms including Gaussian Process Regressor and Efficient Global Optimization predict optimal processing parameters for zero-defect printlets in fused deposition modeling printing. These algorithms achieved impressive prediction accuracies (R² values of 0.8783 for batch printing and 0.9364 for continuous printing) during validation studies.
AI integration with pharmaceutical 3D printing provides multiple benefits. Advanced data-based techniques improve drug stability, dissolution profiles, and bioavailability. Deep learning algorithms predict formulation component interactions under various conditions effectively. This prediction helps determine optimal excipients and processing methods for personalized medicine applications.
Real-World Case Studies from 1000+ AI-Driven Projects

Image Source: Technology Networks
AI’s value in pharmaceutical innovation becomes evident through its ground applications. Case studies from the past several years show how algorithmic approaches deliver measurable results in pharmaceutical companies of all sizes.
Recursion’s Operating System for Rare Disease Discovery
The Recursion Operating System (OS) exemplifies AI-powered drug discovery in action. This complete platform ranges from target identification to clinical trial enrollment and generates 36 petabytes of proprietary data across phenomics, transcriptomics, proteomics, and ADME. Their automated high-throughput labs process up to 2.2 million samples weekly. This capability changes the economics of drug discovery. The company can identify candidate programs within weeks for tens of thousands of dollars, instead of spending millions of dollars over many years through traditional methods. Recursion now has six active clinical-stage programs—four in cancer and two in rare diseases. Seven AI-developed drugs are approaching clinical trial readouts.
Insilico Medicine’s 18-Month IPF Drug Development
Insilico Medicine broke new ground with rentosertib (formerly ISM001-055). They became the first company to find both the biological target and therapeutic compound using generative AI. Their PandaOmics platform analyzed huge datasets to identify TNIK as a novel target for idiopathic pulmonary fibrosis (IPF). Chemistry42, their generative chemistry engine, designed compounds to target this protein. This streamlined workflow helped them progress from target identification to preclinical candidate in just 18 months. Traditional methods typically take more than 10 years. Clinical trials showed promising results – the 60mg dose improved lung function by +98.4 mL, while the placebo group declined by -62.3 mL.
Pfizer’s AI-Optimized COVID-19 Vaccine Manufacturing
Pfizer’s COVID-19 vaccine development showed AI tools’ vital role in manufacturing optimization. Their Smart Data Query (SDQ) tool reviewed clinical trial data within 22 hours after meeting primary efficacy case counts. This innovation cut a month from the development process. Pfizer’s first-in-industry patent-pending Digital Operations Center supported rapid manufacturing scale-up. The center provided complete visibility that helped predict issues and adjust operations immediately. They merged augmented reality for remote equipment diagnosis with IoT sensors for immediate shipment monitoring. This combination maintained cold chain integrity with nearly 100% accuracy worldwide.
Conclusion
AI has revolutionized every phase of pharmaceutical development, as shown by more than 1000 drug discovery projects analyzed in this piece. The old way of discovering drugs took 10 years and $1.4 billion for each successful molecule. AI-driven methods now cut both time and money while boosting success rates at every step.
The numbers tell a compelling story. Deep learning models analyze huge genomic datasets to spot new therapeutic targets with incredible accuracy. Atomwise’s AtomNet platform achieves 74% success rates – this is a big deal as it means that traditional methods are nowhere near as effective. QSAR models and generative networks fine-tune lead compounds precisely and predict toxicity with up to 93% accuracy. AI boosts clinical trials through improved patient stratification and Bayesian optimization, which cuts required participant numbers by up to 71%. Smart formulation design gives optimal drug delivery through excipient compatibility prediction and advanced 3D printing techniques.
These AI technologies have delivered impressive real-life results. Recursion Pharmaceuticals has changed the economics of discovery by identifying candidate programs in weeks rather than years. Insilico Medicine developed an IPF drug in just 18 months – a process that usually takes a decade. Pfizer utilized AI tools to optimize COVID-19 vaccine manufacturing and distribution with nearly 100% cold chain accuracy.
Challenges exist, but the pharmaceutical world has hit a turning point. AI capabilities keep advancing, and we expect faster development of life-changing therapies for patients. Human scientific expertise combined with computational power has created a radical alteration that will deliver better medicines faster and more affordably than ever before.
Key Takeaways
AI is revolutionizing pharmaceutical development by dramatically reducing timelines and costs while improving success rates across every stage of drug discovery.
• AI accelerates target identification by 1-2 years using deep learning to analyze genomic data and protein interactions with 74% success rates versus 50% traditional methods.
• Virtual screening platforms process 15+ quadrillion compounds in days rather than years, identifying novel drug candidates without expensive physical testing.
• Predictive models achieve 93% toxicity prediction accuracy, reducing clinical failures and enabling safer drug development through early risk identification.
• AI-optimized clinical trials reduce participant requirements by 71% through better patient stratification and Bayesian design optimization.
• Real-world results prove AI’s impact: Companies like Insilico Medicine compressed 10-year development cycles to 18 months using AI-discovered targets and compounds.
The pharmaceutical industry has reached a transformative inflection point where AI-human collaboration is delivering life-changing therapies faster, safer, and more cost-effectively than traditional approaches. With the AI pharma market projected to grow from $3.24 billion to $65.83 billion by 2033, this technological revolution is just beginning.